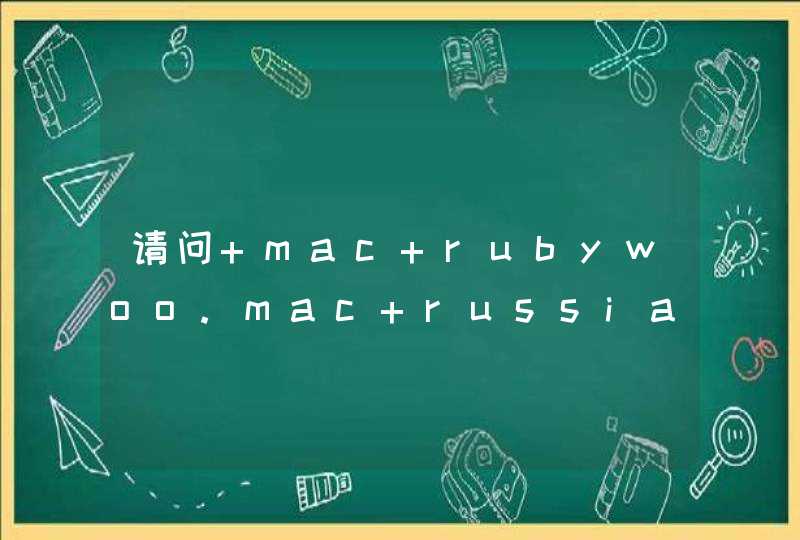## Default S3 method:
cor.test(x, y,
alternative = c("two.sided", "less", "greater"),
method = c("pearson", "kendall", "spearman"),
exact = NULL, conf.level = 0.95, continuity = FALSE, ...)
## S3 method for class 'formula'
cor.test(formula, data, subset, na.action, ...)
根本没有
cor.test(first,second,data= weightBJ_data)
这种调用方式,所以不识别对象first,second
你可以用
attach(weightBJ_data)
cor.test(first,second)
#或者
cor.test(weightBJ_data$first,weightBJ_data$second)
#或者
cor.test(weightBJ_data[,1],weightBJ_data[,2])
你用过attach(weightBJ_data)之后
first才能识别,但应该是没有逗号的。
first[2]
我们输入的数据包含 gene ID 和 vector(单样本)部分,这里的 gene ID 是一个通用概念,可以是基因、转录本、酶或蛋白质。这里的 vector 可以是样本的表达量、倍数变化, p-value, 组蛋白修饰数据等可测量的属性。下面我们以一个 RNA-seq 差异分析后的数据为例,来学习 pathview 的用法。
在 KEGG PATHWAY Database 查询,例如查询小鼠的"Cell Cycle"这条通路:
得到通路 ID 为"04110",物种为"mmu"
我们通过指定 gene.data 和 pathway.id 来观察我们数据里的基因在信号通路“Pathways in cancer”上的表达变化:
相比于原始的 KEGG 图,我们可以使用 graphviz 产生一个新的布局,并且输出 PDF 格式的文件:
以下是输出结果图
如果我们想要运行的更快一点,并且不介意输出图片的大小,我们可以分图层,用 same.layer = F 将节点颜色和标签添加到另一个图层中,并且原来的 KEGG 基因标签会变成官方的 gene symbols :
在此基础上,修改 kegg.native = FALSE ,我们就可以得到一个主图与图例分成两个页面的 PDF 文件
在原始的 KEGG 视图中,一个基因节点可能代表具有相似或者冗余功能的基因/蛋白质,我们可以将这种包含多个基因的节点拆分成独立的节点,这样可以更好的从基因层面而不是节点层面来查看数据。同时也可以通过汇总基因数据来可视化节点数据:
为了画面有更好的清晰度和可读性,默认不分裂节点,也不单独标记每个成员基因。
代谢途径中,除了基因节点还有化合物节点,我们可以尝试利用代谢途径( Propanoate metabolism)整合基因数据和化合物数据。这里的化合物数据包括代谢物、药物,对它们的测量和它们的属性。在这里我们仍然使用之前 RNA-seq 差异分析的数据作为 gene data,然后,我们生成模拟化合物或代谢组数据,并加载适当的化合物 ID 类型以进行演示:
结果如下
pathview 可以集成并将多个样本或状态绘制成一个图,我们可以使用多个重复样本模拟化合物数据:
结果如下,可以看到基因节点和化合物节点被分成多份,对应不同的样本:
我们可以根据将化合物数据分为绝对值大于 5 和小于 5 两类,构成一组离散型数据:
结果如下:
Pathview 包中的主函数是 pathview() ,有着各种参数,是我们用到最多的函数。在这篇文章中,我们介绍了 pathview()的比较常见的用法,包括包安装,数据准备,以及其他有用的特性。我们也可以使用 pathxiew 的网页版,地址是 https://pathview.uncc.edu/ 。此外,Pathview 在数据整合方面有很强大的功能,包含 4800 个物种,能处理的数据属性和格式包括 连续/离散数据、矩阵/矢量、单个/多个样本数据 ,包中还具有强大的 ID 转换功能,这些都值得我们进一步探索。
生活很好,有你更好
先加载相关的package
然后提取想要的基因集,变成list
然后进行富集分析
需要注意的点:
1.expr输入的表达矩阵必须为:SummarizedExperiment或者SingleCellExperiment ExpressionSet 或者别的什么对象。如果是dataframe的话需要转换为matrix。
2.gset.idx.list是输入的基因集,要么是一个list,要么是一个GeneSetCollection对象
3.kcdf这个参数用于设定非参数检验的分布类型,当输入的表达矩阵是raw_counts时,那么这个参数应该设置为kcdf=“Poisson”。
颜色通过colorRampPalette(c("navy", "white", "firebrick3"))(100)函数生成
annotation_col是一个1列的dataframe,行名是样本名,第一列填分组信息,要转换为factor。如下图:
得到的热图如下图所示: