

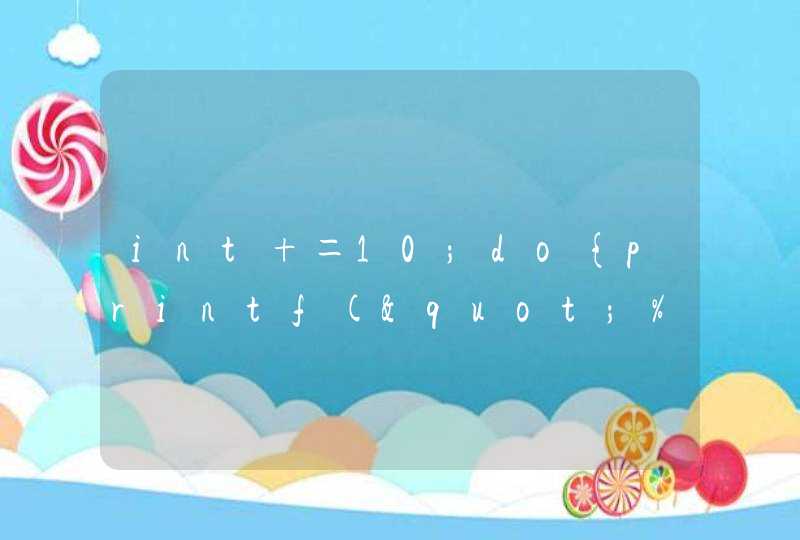






































1 读取,计算均值,箱图观察
2 查看数据分布
2.1 hist直方图
2.2 qqnorm散点图
3 Shapiro-Wilk正态性检验
4 方差齐性检验
意义:方差分析就是在大家误差水平差不多的条件下看控制和对照组是不是有显著差异。那方差其实就是误差水平了。当方差不一致的时候,这个方法就没法分辨出究竟是控制造成的差异还是,内在的波动造成的差异。
参考: https://www.zhihu.com/question/21195390
参考: https://blog.csdn.net/tiaaaaa/article/details/58130363
4.1 F检验
使用条件:数据正态分布,只可以检验两个样本
4.2 bartlett检验
使用条件:正态分布的数据,多个样本
4.3 levene检验
没有条件:数据可不具正态性,可以检验多个总体的方差齐性
SPSS的默认方差齐性检验方法
5 差异检验
5.1 参数检验:T检验
使用条件:两样本来自正太分布总体,方差齐
5.2 非参数检验:Wilcoxon秩和检验(两样本)
参数:
参考: https://www.jianshu.com/p/f30d1fe877ea
5.3 非参数检验:Kruskal-Wallis(KS)秩和检验(多样本)
5.4 Deseq两组reads count差异分析
#mRNA表达矩阵与GROUP文件样式,heatmap样式见文章最后
library(limma)
mRNA <- read.table("表达矩阵.txt",sep = "\t",header = T,comment.char = "!",encoding = "UTF-8")
#mRNA数据框行名为基因名,列命为样本名称
group <- read.table("GROUP.txt",header=T,sep = "\t",encoding = "UTF-8")
group_CP <- group$treat
m_design<- model.matrix(~0+factor(group_CP))
colnames(m_design) = levels(factor(group_CP))
rownames(m_design)= group$ID
contrast.matrix<-makeContrasts("P-C",levels=m_design) #注意P-C顺序,实验组要在前面否则影响上下调结果
m_fit <- lmFit(mRNA,m_design)
m_fit <- contrasts.fit(m_fit, contrast.matrix)
m_fit <- eBayes(m_fit)
m_genlist <- topTable(m_fit, coef = 1, n=Inf) #limma结果
#将表达矩阵与差异分析结果合并
ID_REF <- rownames(m_genlist)
m_genlist <- data.frame(ID_REF,m_genlist)
ID_REF <- rownames(mRNA)
mRNA <- data.frame(ID_REF,mRNA)
test <-merge(mRNA,m_genlist,by = "ID_REF")
result <- subset(test,P.Value<0.05)
row.names(result) <- result[,1]
#绘制热图
heatmap <- result[2:(nrow(group)+1)]
annotation <- data.frame(Factor = factor(group$treat)) #标注样本的分组信息
rownames(annotation) <- colnames(heatmap)
library(pheatmap)
filename <- paste("文件名",".pdf",sep="")
pdf(filename)
pheatmap(heatmap,
annotation=annotation,
annotation_legend = TRUE,
main=filename ,
scale = "row",
show_rownames = F,
color = colorRampPalette(c("green","black","red"))(100))
dev.off()
#表达矩阵与GROUP文件如下所示





























