















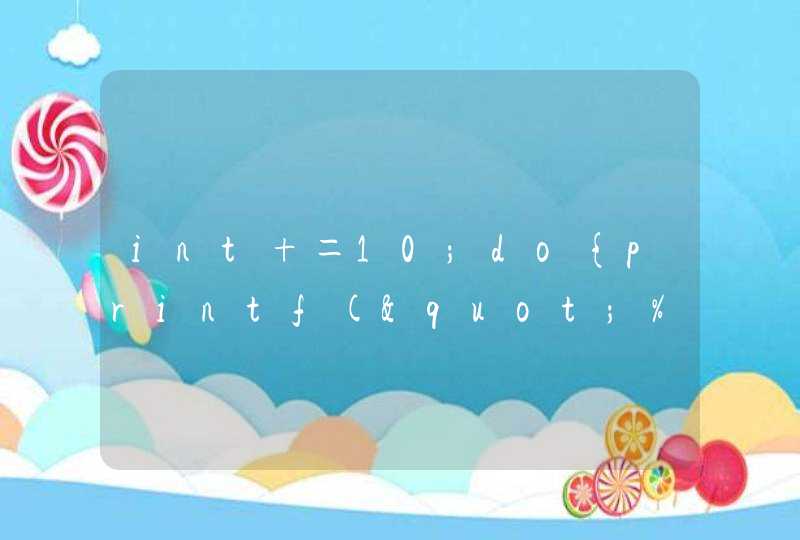
























TCGA里的数据tumor多normal少,想要挑选出配对样本进行差异分析。(并不是说必须挑配对样本才能做哦,直接做也是可以的)
这里使用的数据是TCGA的CHOL表达矩阵。
从TCGA ID里可以找到tumor和normal的分组,统计它们的数量:
配对样本的共同点是病人ID(前12位)一致,根据这个来匹配即可。
先拆分成tumor和normal两个矩阵,根据ID的14和15位来拆
有normal样本的病人的ID ,是normal组样本ID的前12位,也是exp_nor的行名。
然后将tumor矩阵中与patient相匹配的样本名选出来,能匹配到的就会被挑出来。%in%从来都是R语言里的一个神技能: %in%很简单
可以把表达矩阵拼回去咯:
就相当于把表达矩阵按列取子集了,tumor和normal各九个样本,后面可以拿来做配对样本的差异分析哦,例如下面这张图,就是COAD数据配对差异分析得到的差异基因热图。
转录组的差异分析在这里: TCGA(转录组)差异分析三大R包及其结果对比
获取表达矩阵,处理TCGA的count数据,1表示为行。
导入数据
加 ENTREZID列,用于富集分析(symbol转entrezid,然后inner_join)
转化空格为NA
用花花的专属TCGA包,ID进行转换
把空着的值改为NA
以病人为中心,表达矩阵按病人ID去重复
去除重复
TPM数据做单个基因的生存分析file:///C:/Users/denghuan/Desktop/The%20learning%20of%20R%20software/Practice/%E7%94%9F%E5%AD%98%E5%88%86%E6%9E%90%20survival%20analysis/6.Survival.html
stringr::str_replace_all()
str_detect(colnames(exp),"TCGA-W5-AA2R")





























