









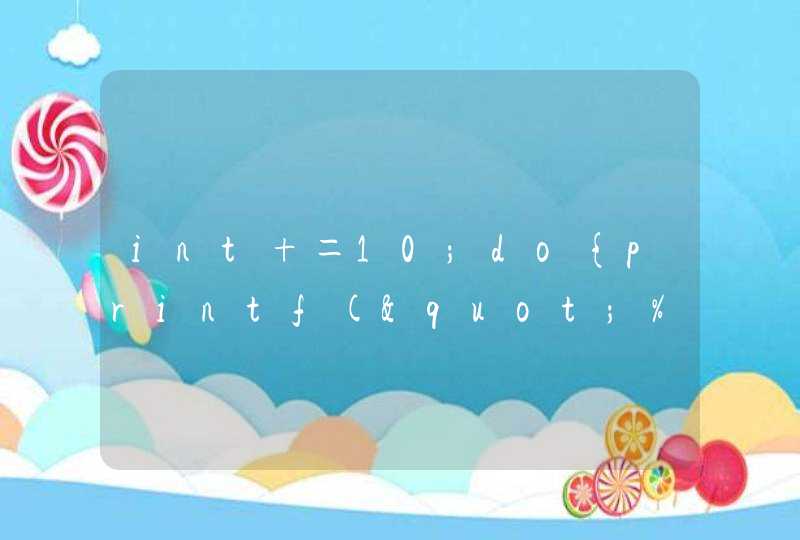






























微生物群落测序是指对微生物群体进行高通量测序,通过分析测序序列的构成分析特定环境中微生物群体的构成情况或基因的组成以及功能。借助不同环境下微生物群落的构成差异分析我们可以分析微生物与环境因素或宿主之间的关系,寻找标志性菌群或特定功能的基因。对微生物群落进行测序包括两类,一类是通过16s rDNA,18s rDNA,ITS区域进行扩增测序分析微生物的群体构成和多样性;还有一类是宏基因组测序,是不经过分离培养微生物,而对所有微生物DNA进行测序,从而分析微生物群落构成,基因构成,挖掘有应用价值的基因资源。以16s rDNA扩增进行测序分析主要用于微生物群落多样性和构成的分析,目前的生物信息学分析也可以基于16s rDNA的测序对微生物群落的基因构成和代谢途径进行预测分析,大大拓展了我们对于环境微生物的微生态认知。目前我们根据16s的测序数据可以将微生物群落分类到种(species)(一般只能对部分菌进行种的鉴定),甚至对亚种级别进行分析,几个概念:16S rDNA(或16S rRNA):16S rRNA 基因是编码原核生物核糖体小亚基的基因,长度约为1542bp,其分子大小适中,突变率小,是细菌系统分类学研究中最常用和最有用的标志。16S rRNA基因序列包括9个可变区和10个保守区,保守区序列反映了物种间的亲缘关系,而可变区序列则能体现物种间的差异。16S rRNA基因测序以细菌16S rRNA基因测序为主,核心是研究样品中的物种分类、物种丰度以及系统进化。OTU:operational taxonomic units (OTUs)在微生物的免培养分析中经常用到,通过提取样品的总基因组DNA,利用16S rRNA或ITS的通用引物进行PCR扩增,通过测序以后就可以分析样品中的微生物多样性,那怎么区分这些不同的序列呢,这个时候就需要引入operational taxonomic units,一般情况下,如果序列之间,比如不同的 16S rRNA序列的相似性高于97%就可以把它定义为一个OTU,每个OTU对应于一个不同的16S rRNA序列,也就是每个OTU对应于一个不同的细菌(微生物)种。通过OTU分析,就可以知道样品中的微生物多样性和不同微生物的丰度。测序区段:由于16s rDNA较长(1.5kb),我们只能对其中经常变化的区域也就是可变区进行测序。16s rDNA包含有9个可变区,分别是v1-v9。一般我们对v3-v4双可变区域进行扩增和测序,也有对v1-v3区进行扩增测序。
测序行业的蓬勃发展,带来微生物组学日新月异的变化。目前,单一组学的文章不断“贬值”,前沿研究的目光从单一组学逐步拓展至多组学对贯穿分析,即结合多个组学的分析角度,从多个层面阐述生物学机制。
微生物多组学贯穿分析策略十分丰富:如常见的16s与宏基因组贯穿分析,可以验证物种的特征、丰富功能的探究;而16s与代谢组的贯穿分析思路同样常见于高分文章中,通过16s探究不同处理/环境下菌群的物种组成变化,结合代谢组对应的代谢物的变化,进而找到不同处理/环境下引发细菌丰度差异最终导致代谢表型差异的机制。参考阅读《选好思路和方法,给自己一篇多组学高分文章 》
在16s与代谢组贯穿分析中,相关性热图是一个重要的分析手段,主要用于逐一呈现细菌物种与代谢物间的相关性高低,是筛选潜在关联的物种与代谢物的主要途径,对于下游的实验起到指导意义。此类相关性热图在高分文章中频繁出现,足见其重要性(图1、图2)。
图1 物种代谢物热图(2015,Cell Host&Microbe,IF=15.753 )[1]
图2 物种代谢物热图(2018,NatureMedicine,IF=30.641)[2]
那么,该如何画出此类高分文章中的相关性热图呢?这里,以16s与代谢组的数据为例,向大家分享如何使用R语言进行两个组学数据的相关性计算、绘制相关性热图。
1.加载R包
library(psych)
library(pheatmap)
library(reshape2)
2.读入数据
phy <-read.table(file = "phy.xls", sep = "t", header = T,row.names= 1)
图3 微生物丰度信息表格
met <-read.table(file = "met.xls", sep = "t", header = T,row.names= 1)
图4 代谢物丰度信息表格
3.计算相关性、p值
cor <-corr.test(phy, met, method = "pearson",adjust= "none")
cmt <-cor$r
pmt <- cor$p
head(cmt)
head(pmt)
4.数据保存
cmt.out<-cbind(rownames(cmt),cmt)
write.table(cmt.out,file= "cor.txt",sep= "t",row.names=F)
图5 相关性系数表格
pmt.out<-cbind(rownames(pmt),pmt)
write.table(pmt.out,file= "pvalue.txt",sep= "t",row.names=F)
图6 p值表格
df <-melt(cmt,value.name= "cor")
df$pvalue <- as.vector(pmt)
head(df)
write.table(df,file= "cor-p.txt",sep= "t")
图7 关系对信息
5.绘制显著性标记
if(!is.null(pmt)){
ssmt <- pmt<0.01
pmt[ssmt] <- '**'
smt <- pmt >0.01&pmt <0.05
pmt[smt] <- '*'
pmt[!ssmt&!smt]<- ''
} else{
pmt <- F
}
6.绘制相关性热图
mycol<-colorRampPalette(c("blue","white","tomato"))(800)
pheatmap(cmt,scale = "none",cluster_row = T, cluster_col = T, border=NA,
display_numbers = pmt,fontsize_number = 12, number_color = "white",
cellwidth = 20, cellheight =20,color=mycol)
图8 R语言绘制的物种+代谢物相关性热图
pheatmap(cmt,scale = "none",cluster_row = T, cluster_col = T, border=NA,
display_numbers = pmt, fontsize_number = 12, number_color = "white",
cellwidth = 20, cellheight = 20,color=mycol,filename= "heatmap.pdf")
参考文献
[1]Kostic AD, Gevers D, Siljander H, et al. The dynamics ofthe human infant gut microbiome in development and in progression toward type 1diabetes. Cell Host Microbe. 201517(2):260–273.doi:10.1016/j.chom.2015.01.001
[2]Hoyles, Lesleyet al. “Molecular phenomics and metagenomics of hepatic steatosis innon-diabetic obese women.” Nature medicine vol. 24,7 (2018):1070-1080. doi:10.1038/s41591-018-0061-3
原文 https://www.sohu.com/a/366652239_278730
用limma包,这里注意,limma包是对基因芯片表达矩阵的分析,不能对逆转录RNAseq表达矩阵进行分析(因为数据特征不同),RNAseq需要用另一种方法
解读此表
但是上面的用法做不到随心所欲的指定任意两组进行比较,所有还有下一种方法
处理好了分组信息,再自定义比较元素
自定义函数进行比较
热土和火山图都是傻瓜式的,只要的前面得出的deg数据(也就是基因差异表达数据)是正确的





























