






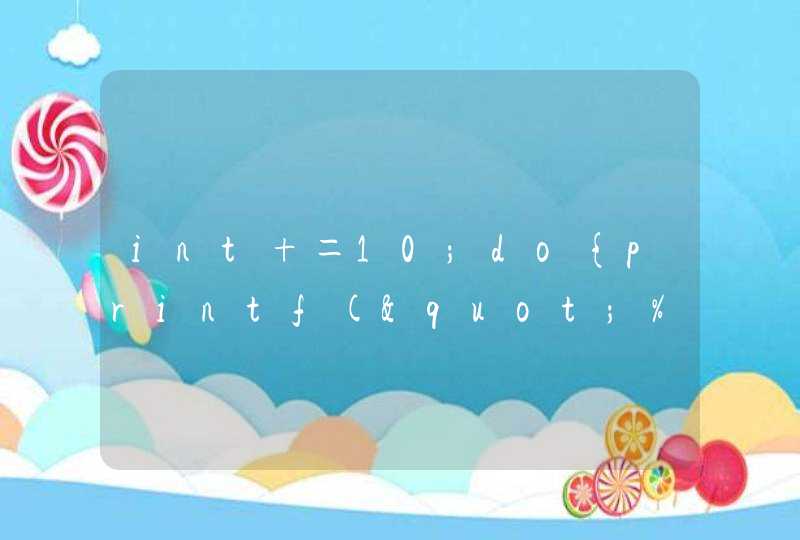
































,第1张")
字符串拼接函数 str_c ,与R语言自带的 paste 和 paste0 函数具有相同的作用。
字符计数函数 str_count ,计算字符串中指定字符的个数。
字符检查函数 str_detect ,检查字符串中是否包含指定字符,返回逻辑向量。
字符复制函数 str_dup ,将字符向量重复若干次,返回重复后的字符向量。
字符提取函数 str_extract 和 str_extract_all ,对字符串进行提取, str_extract_all 函数返回所有的匹配结果。
字符串格式化函数 str_glue ,用花括号 {} 表示占位符,括号内的变量被替换成全局变量值。
字符串长度函数 str_length ,计算字符串长度。
字符位置提取函数 str_locate 和 str_locate_all ,返回匹配到的字符的位置。
字符匹配函数 str_match 和 str_match_all 与字符提取函数 str_extract 类似,返回匹配到的字符,不同之处在于返回格式。
字符补齐函数 str_pad ,用于在字符串中添加单个字符,可选择添加的位置,在参数 side 中进行设置。
字符删除函数 str_remove 和 str_remove_all ,用于删除字符串中的部分字符。
字符替换函数 str_replace 、 str_replace_all 和 str_replace_na ,用于替换字符串中的部分字符。
字符排序函数 str_sort 和 str_order ,对字符向量进行排序。
字符分割函数 str_split 和 str_split_fixed ,对字符串进行分割。
字符过滤函数 str_sub 和 str_subset , str_sub 函数通过指定开始和结束位置,过滤出字符串的部分字符串。 str_subset 函数通过匹配模式,过滤出满足模式的字符串。
stringr 包中其他的有用函数,用于常见的字符处理。
目录
vcf数据包含了所有的等位对立基因的信息,这样就可以帮助我们判断染色体的倍数。比方说有一个位点的碱基是A/T,测序覆盖率为20, 如果这个物种是二倍体,那么A,T的出现概率就是(50%),会各自出现10次,如果是3倍体,那么A会出现13次,T会出现7次,当然也有可能相反。当把所有的点位集合在一起的时候,我们就可以判断这个物种的倍数体了。
用包里的自带数据,有疑问的小盆友可以查阅之前的文章,这里就不做赘述了。
高通量数据测序可以保证每一个位点都经过很多次的读取,这样就相当于每一个等位基因都被测序过了差不多相等的次数。假设我们对一个二倍杂合体进行了覆盖率为30的测序,那么每一条染色体都被测了15次。当然真实情况不可能正好是这个数字,毕竟测序的时候会发生一定概率的错误。
假设我们用覆盖率为30给一个三倍杂合体进行测序,某基因位点为A/A/T,那么,A和T出现的期待值将是20和10。当某个基因位点的组合是A/G/C时,那么A,G,C就会各自出现10次。
FORAMT里的AD表示对立基因的各自出现的次数。所以我们可以提取AD数据。
一般的SNP Caller都会默认双倍体检验,也就是出现两种对立基因型。所以可以计算每种基因的出现概率。
然后用直方图可视化一下。
可以发现,大多数都是纯合,所以需要去掉纯合的部分。
我们发现峰值出现在了1/2,说明这个物种时二倍体,和预期的一样。
然而这里有一个小小的问题,Fequency几乎从0到1横跨整个横坐标,这个明显不合理,需要进行改善。
我们可以通过等位对立深度(AD)的信息来改善刚才提到的问题。
我们可以看到80%的数据分布在了19和75之间。然后再靠近40和60点的地方出现了两个峰,这分别代表杂合峰和纯合峰。然后整个数据还拖着一个尾巴,最长的地方超过了100,这表示部分区域包含了着非常高的拷贝数(CNVs)。此处的目的是为了可视化倍数体,所以选择100以下15%~95%的数据。
回想一下之前文章里介绍过的用箱图做可视化的内容,我们也可以通过同样的方法来确认过滤数据的效果。
看一下过滤后的结果。
果然好看很多。
最后再回到一开始,看倍数体的可视化效果。
结果明显干净易懂好多。
有同学会问,那么不是二倍体的话会出现什么样的结果呢。数据包的样本里正好有一个三倍体。
可以看到两个峰出现在了1/3,2/3处。结果和实际完美匹配。





























